Mitochondrion
n., plural: mitochondria
[ˌmaɪtəʊˈkɒndrɪən]
Definition: a semi-autononomous organelle where cellular respiration occurs
Table of Contents
Mitochondrion Definition
What are mitochondria? The term “mitochondrion” comes from the two words of the Greek language “mitos” and “chondrios” meaning “thread” and “grain” respectively. The term “mitochondrion” is singular whereas the word “mitochondria” is plural. As we have long known, the mitochondria are the powerhouse of the cell. They provide chemical energy to the cell in the form of ATP molecules. The definition of mitochondria in biology goes like this:
Mitochondria are round or oval shaped, double-membrane-bound organelles in the eukaryotic cells that pull off the responsibility of keeping a cell charged to do cellular work by the production of energy units called ATP.
What does a mitochondrion do?
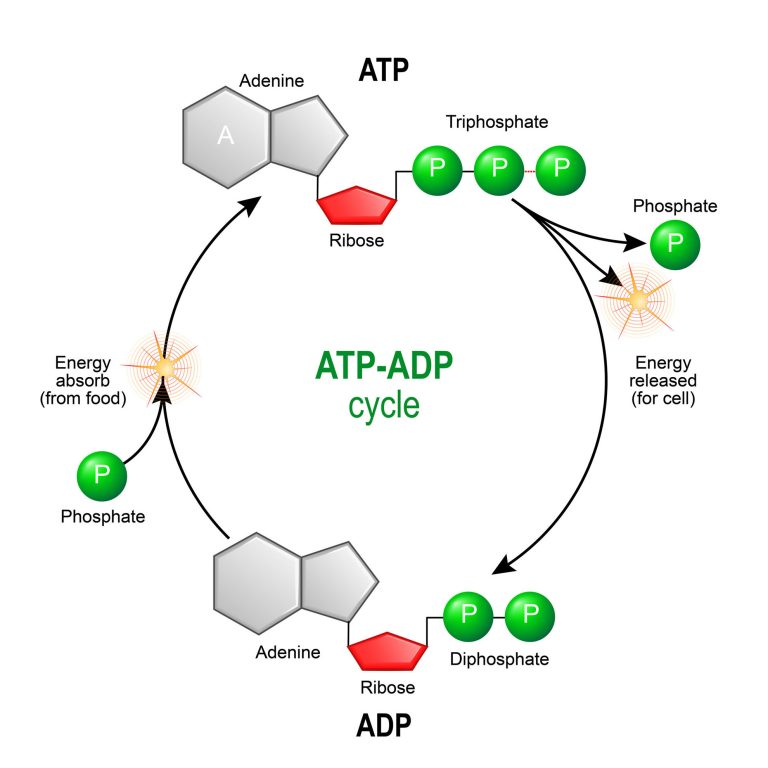
The mitochondrion is the organelle of a cell that is actively involved in the production of energy or fuel for the basic biological functioning of a cell. Now the question arises, what is ATP? ATP – or adenosine triphosphate – is a high-energy compound that fuels cellular energy requirements. It is produced by the process of cellular respiration that takes place in the mitochondrion. During cellular respiration the food is oxidized, oxygen is consumed, and carbon dioxide is released.
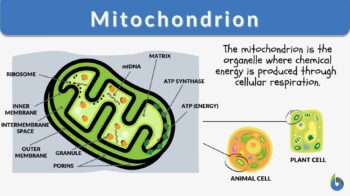
The mitochondrion is a spherical or rod-shaped organelle that has its own genome and is responsible for the generation of most of the cell’s supply of adenosine triphosphate through the process of cellular respiration. This is why mitochondrion is regarded as the powerhouse of eukaryotic cells. Etymology: from Greek “mitos”, meaning “thread” and “khondrion”, meaning “little granule”. Synonym: chondriosome. Related form: mitochondrial (adjective, of, or relating to, mitochondrion). See also:
Now that we’re well-versed with the basics and know how to define mitochondria, let’s move forward and try to decipher the origin and evolution of this organelle…
Origin and Evolution of Mitochondria
Before we begin to learn the theories and hypotheses associated with the origin of mitochondria, let’s answer some basic questions and queries related to this amazing organelle.
- Do plants/plant cells have mitochondria?
Answer: Yes, since plants are eukaryotes and all eukaryotic cells (generally) possess mitochondria, so plant cells also have mitochondria. - Do prokaryotes have mitochondria?
Answer: No, prokaryotes don’t possess mitochondria. - Do bacteria have mitochondria?
Answer: No, Bacteria are prokaryotic organisms and therefore they don’t have any mitochondria. - Are mitochondria present in plant and animal cells?
Answer: Yes, both plant and animal cells (in general) have mitochondria.
As a matter of fact, mitochondria are found in most eukaryotic cells. The exact origin & evolution of the mitochondria remains a mystery to humankind. However, there are two hypotheses that try to explain the origin of mitochondria.
1. Endosymbiotic hypothesis
This is, by far, the most widely accepted hypothesis. According to it, the present-day mitochondria originate from a free-living aerobic prokaryotic organism, possibly an α-proteobacteria. The ancestral pre-eukaryotic cell engulfed the bacterium which survived the endocytosis and developed a symbiotic relationship. Over the course of evolution, the engulfed bacterium lost its cell wall & most of the DNA which was not useful for the host cell. The hypothesis is supported by shared similarities between bacteria and mitochondria.
Some striking similarities between mitochondria and bacteria:
- Presence of circular DNA in both bacteria and mitochondria.
- The 70S ribosomes of mitochondria and bacteria are similar in size and structure.
- Presence of porins in the outer mitochondrial membrane and bacterial cell membrane.
- Presence of cardiolipin in the inner mitochondrial membrane and bacterial cell membrane.
- New mitochondria are formed only by binary fission and cannot be produced by the cell.
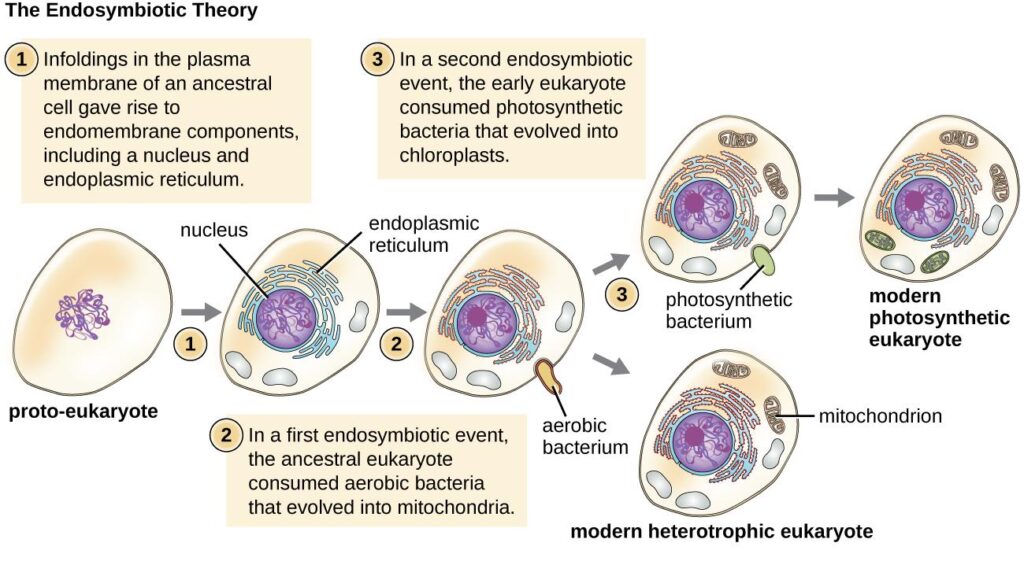
2. Autogenous hypothesis
According to this theory, the present-day mitochondria were formed by the functional clustering of the DNA from the main genome and subsequent compartmentalization due to the invagination of the cell membrane.
The mitochondria might have originated from early bacteria that became so symbiotic with their hosts, the eukaryotic cells, that they evolved and become indispensable energy-yielding structures within the eukaryotic cells (endosymbiotic theory). Nevertheless, there is a eukaryote that lacks mitochondrion. Monocercomonoides is the first eukaryotic species found to be devoid of mitochondria. It obtains energy by metabolizing nutrients absorbed from the environment.READ: Prokaryotic Ancestor of Mitochondria: on the hunt
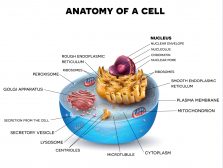
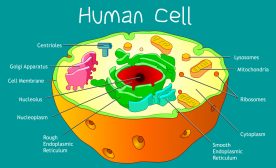
Mitochondria Structure
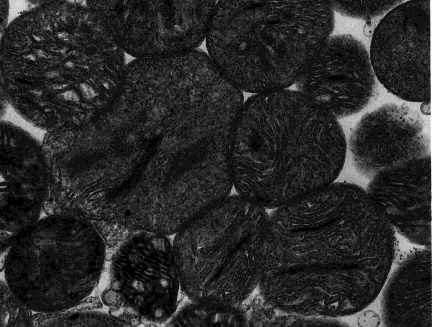
A mitochondrion is a tiny organelle that is typically round to oval and 0.75-3 μm² in size. It is a double membrane-bound organelle found in the cytoplasm of most eukaryotic cells. The dual membranes make the mitochondria similar to the other organelles such as the nucleus and plastids. The number and shape can vary greatly depending on the type of cell.
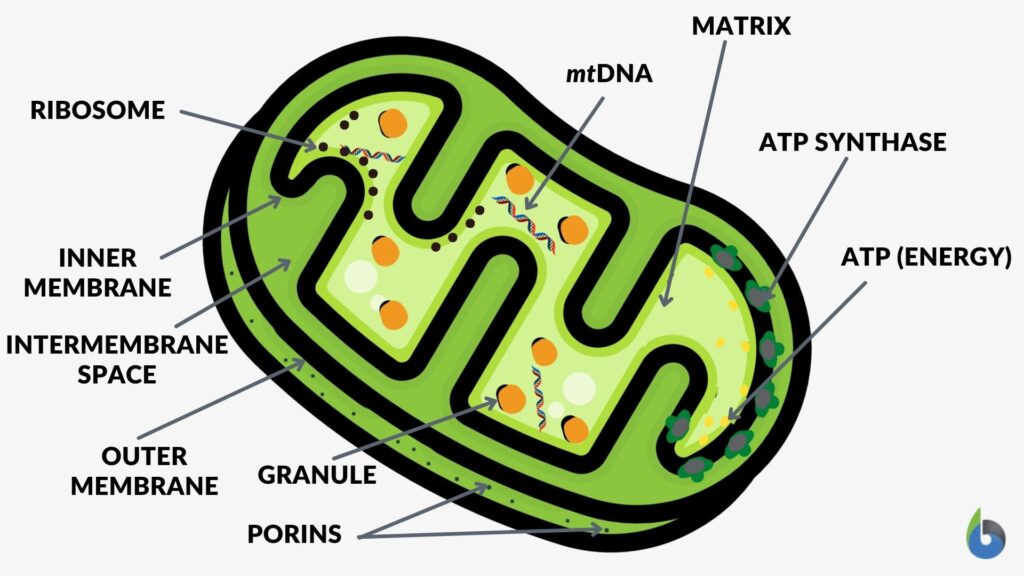
The structure of the mitochondria can be broken down into five major components. The list below describes the different parts of mitochondria.
1. Outer membrane
The outer membrane is the outermost covering of the mitochondrion. It separates the contents of intermembrane space from the cytosol. It is like a plasma membrane in its composition as both are composed of phospholipid bilayer & contain many embedded proteins and enzymes. It is permeable to small molecules of <1000 kDa. It has a major regulating role in programmed cell death.
List of important components of the outer membrane of mitochondria along with their significance:
- Porins: specialized protein structures that allow the passage of ions, nucleotides, metabolites, and small proteins across the membrane.
- Translocase: they are involved in the transport of some large proteins.
- Metabolic enzymes: Mono-amine oxidase, Kynurenine hydroxylase, Fatty-acid CoA ligase, NADH-Cyt-c reductase.
- MAM (Mitochondria-associated ER-membrane): MAM are the functional domains that connect mitochondria & ER. They play a role in the transport of lipids, calcium homeostasis, autophagy, and apoptosis.
2. Intermembrane space
The intermembrane space – or peri mitochondrial space — is a tiny space present between the outer and the inner mitochondrial membrane. It plays a crucial role in the transport of proteins & ions, assembly of inner membrane proteins, and cellular respiration. It houses the protons that are pumped from the mitochondrial matrix into the intermembrane space during the redox reactions of the electron transport chain.
3. Inner mitochondrial membrane
The inner membrane of mitochondria is larger in size than the outer membrane. It creates numerous folds in order to accommodate itself within the outer membrane thus giving it a wrinkled appearance when observed under an electron microscope. It is the primary site of oxidative phosphorylation as it contains the complexes of the electron transport chain. Unlike the outer mitochondrial membrane, the inner mitochondrial membrane is highly impermeable and is devoid of porins. This enables the inner mitochondrial membrane to maintain the proton gradient that drives the ATP synthesis.
4. Cristae
Numerous folds of the inner mitochondrial membrane create compartments which are known as cristae. These cristae are studded with proteins of the electron transport chain & help in maximizing the surface area for reactions of oxidative phosphorylation. Cells with high requirements of ATP/energy have more cristae in their mitochondria.
5. Matrix
The space enclosed within the bounds of the inner mitochondrial membrane is the matrix of the mitochondria. It houses several copies of mitochondrial DNA & various metabolic enzymes. One of the most vital metabolic cycles i.e., Krebs cycle (also called TCA cycle) occurs in the mitochondrial matrix.
Structure of the Mitochondrion:
The mitochondrion consists of outer and inner membranes, an intermembrane space (space in between the membranes), the cristae (infoldings of the inner membrane), and the matrix (space within the inner membrane). The outer membrane contains several porins that form channels where certain molecules can freely diffuse. Unlike the outer membrane, the inner membrane does not contain porins and is highly impermeable to all molecules. Most ions and molecules would need special membrane transporters to enter or exit the mitochondrial matrix. The cristae, which are the foldings of the inner membrane, increase the surface area, increasing ATP production. The matrix contains enzymes, mitoribosomes, tRNA, and DNA.
Mitochondrial DNA
Mitochondria are unique in their existence as they contain their own physically and transcriptionally separate genome. There are chiefly six different types of mitochondrial DNA that exist across various species. Human beings contain circular mitochondrial DNA whereas certain plants, fungi, and protists contain linear forms of DNA. Humans and most animal mitochondria have smaller genomes of around 16 kb. Yeast and plants have significantly larger mitochondrial genomes of 80 kb and 200 kb respectively. However, these larger genomes don’t necessarily contain more genetic information.
The list below mentions the salient features of the human mitochondrial DNA
- It consists of 16,569 base pairs.
- It has a total of 37 genes.
- 13 genes code exclusively for proteins involved in oxidative phosphorylation
- 2 genes code for rRNA: 16S & 12S rRNA
- 22 genes code for tRNA
Mitochondrial DNA is strikingly different from nuclear DNA. The key differences are tabulated below.
Table 1: Differences between the mitochondrial and nuclear DNA in humans |
|
|---|---|
| Mitochondrial DNA | Nuclear DNA |
| Circular | Linear |
| 16,569 base pairs | 3 billion base pairs |
| Multiple copies in a single cell | One copy in a single cell |
| Non enveloped | Enveloped |
| Open DNA | DNA packed in chromatin |
| Dependent on certain nuclear DNA gene products | Independent of mitochondrial DNA |
| Does not follow the universal genetic code | Follows the universal genetic code |
| Genes are overlapping | Genes are not overlapping |
| Very few noncoding regions | 93% of DNA is noncoding |
| Maternal inheritance | Both maternal and paternal inheritance |
| No involvement of histone proteins; mtDNA lies histone-free just like nucleoid of bacterial cells. | Histone proteins are involved in the packaging of nuclear DNA into nucleosomes. |
Transmission of mitochondrial DNA
It is believed that in most organisms including humans, mitochondrial DNA is exclusively inherited from the mother. However, according to a study, there have been rare instances of heteroplasmy of mitochondrial DNA in humans, which points that exceptionally the paternal mitochondrial DNA may also contribute.
Due to this exclusive maternal inheritance, it is possible to trace human maternal lineage to the mitochondrial Eve, mother, or female biological ancestor of all human beings. Paternal inheritance of mitochondrial DNA has been observed in some other species as well but their exact role and significance are debatable.
Mitochondrial DNA:
The mitochondrial DNA is genetically distinct from that in the nucleus. Since a mitochondrion has its own genetic material and is capable of manufacturing its own RNAs and proteins, it is said to be a semi-autonomous, self-reproducing structure. In fact, mitochondrial DNA has become an important tool in tracking genetic histories since the genetic material is present in only one copy, and does not recombine during reproduction.
READ:
Transcription and Replication
Transcription and replication of mitochondrial DNA are distinct from nuclear DNA. Mitochondrial DNA encodes for most of its own replication machinery. The replication of the mitochondrial DNA is carried out by its own special DNA polymerase gamma complex encoded by the POLG gene. Unlike nuclear DNA transcription, mitochondrial DNA transcription does not follow the genetic code. The table below highlights the “violations” of the genetic code by mitochondrial DNA. Some examples of mitochondria codon variations from universal codons can be seen here.
Table 2: “violations” of the genetic code by mitochondrial DNA |
||
|---|---|---|
| Codon | Universal Code | Mitochondrial DNA |
| UGA | Stop codon | Tryptophan |
| AUA | Isoleucine | Methionine |
| AGA/AGG | Arginine | Stop codon |
Mitochondria Function
Now let’s answer the question, what is the purpose of the mitochondria? Mitochondria have a role in many important functions of the cell some of which are mentioned in the list below.
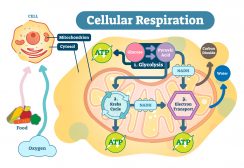
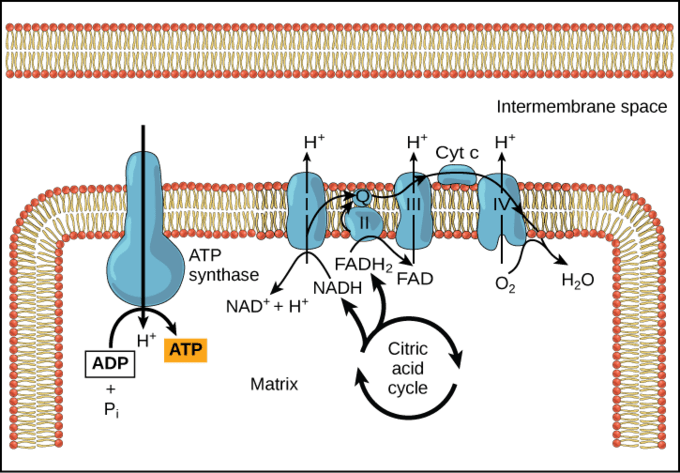
- Synthesis of ATP – The most vital function of the mitochondrion is the synthesis of ATP. ATP is the energy currency of the cell that powers the biochemical reactions. ATP molecules are produced by a process called oxidative phosphorylation. Oxidative phosphorylation is a series of complex redox reactions in the electron transport chain which oxidizes the nutrient metabolites thereby releasing energy that is utilized for phosphorylation of ADP to ATP.
The electron transport chain is located in the inner mitochondrial membrane. The electron transport chain is a series of complexes arranged in increasing order of redox potential that act as electron acceptors. Molecules like NADH and FADH2 donate electrons via redox reactions to the complexes of the electron transport chain. These redox reactions are coupled with the pumping of protons into the intermembrane space thus creating an electrochemical gradient across the inner mitochondrial membrane. This gradient is used by the FOF1 ATP synthase complex to synthesize ATP.
Figure 6: ATP synthesis. Image Credit: LibreTexts. - Production of heat – the mitochondrial electrochemical gradient generated by the electron transport chain is utilized for the phosphorylation of ADP to ATP. This is known as chemiosmotic coupling. Thus, the energy released during the electron transfer is stored in high-energy phosphate bonds. However, tissues such as brown fat display no chemiosmotic coupling. Hence, the free energy is released as such in the form of heat. This heat energy helps small children to keep themselves warm as they lack other mechanisms of heat generation such as shivering.
- Homeostasis of calcium – Mitochondria play a vital role in the homeostasis of calcium. Mitochondria and endoplasmic reticulum are the major sites of intracellular calcium stores. MAM are the domains between mitochondria and endoplasmic reticulum tightly regulating cellular calcium levels, which is essential for the cell’s survival & functioning.
- Regulation of Immune function – Mitochondria play a role in the activation, survival, and differentiation of immune cells. It can do so by creating alterations in the metabolism, activating the inflammatory response, altering the mitochondrial dynamics by mitochondrial fission and fusion, and mitochondrial-ER junctional signaling.
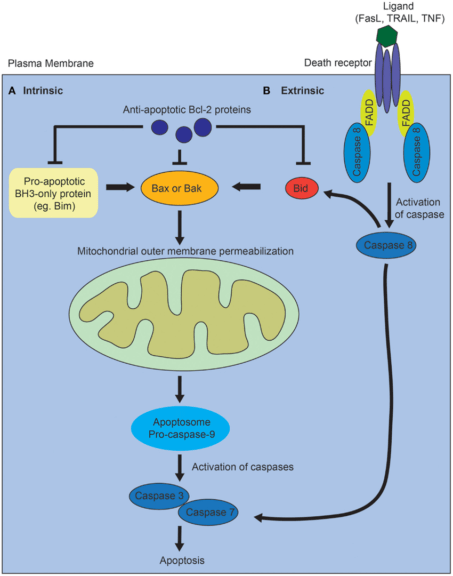
- Apoptosis or programmed cell death – Mitochondria has a well-established role in the intrinsic pro-apoptotic pathway. Cues, such as damage to the DNA, trigger the intrinsic apoptotic pathway. It precedes an imbalance between pro-apoptotic and anti-apoptotic proteins. This imbalance leads to increased permeability of the outer mitochondrial membrane to cytochrome-c (cyt-c), which leaks out in the cytosol. Cyt-c forms complex with APAF-1 (apoptosis activating factor), which recruits and activates caspase-9 (initiator protease). Caspase-9 further activates caspases-3, 6, and 7 (executioner protease). This results in cell death and efficient phagocytosis of the remains.

Figure 7: Mechanism of programmed cell death via intrinsic and extrinsic pathways. Image Credit: Ho J. (2014). - Stem Cell Regulation – Mitochondrial metabolism is vital for the tight regulation of homeostasis of somatic stem cells (SCC). Mutations in mitochondrial DNA result in mitochondrial dysfunction and compromised homeostasis of SCC. This is linked with increased tissue degeneration and aging.
- Synthesis of biochemicals – Mitochondria provides intermediates for the synthesis of several compounds like amino acids, chlorophyll, cytochromes, steroids, alkaloids, pyrimidines, etc. The intermediates of the TCA cycle act as precursors of the synthesis of certain amino acids. Oxaloacetate and α -ketoglutaric acid are precursors of aspartic acid and glutamic acid respectively.
Organization and Distribution
Mitochondria are tiny organelles that are not visible under normal microscopy unless stained specifically. The number, size, and shape of mitochondria in a cell can vary greatly depending on the type of organism, type of cell, and function of a cell. They are dynamic organelles that can rapidly undergo cycles of fusion and fission in order to adapt to cellular requirements. When two different mitochondria fuse together to become one it is called fusion and when a mitochondrion splits into two it is called fission. The total mitochondria population of the cell is termed chondriome. They often form complex interconnected networks with the cytoskeleton of the cell. They are always distributed along with the microtubules and often associated with the endoplasmic reticulum.
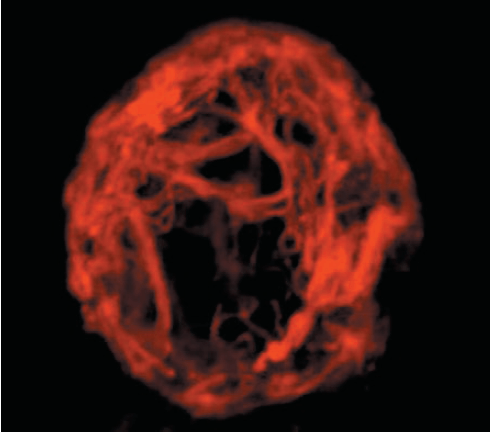
The diversity of structure & distribution of the mitochondria across various organisms & cell types can be understood by the examples provided below:
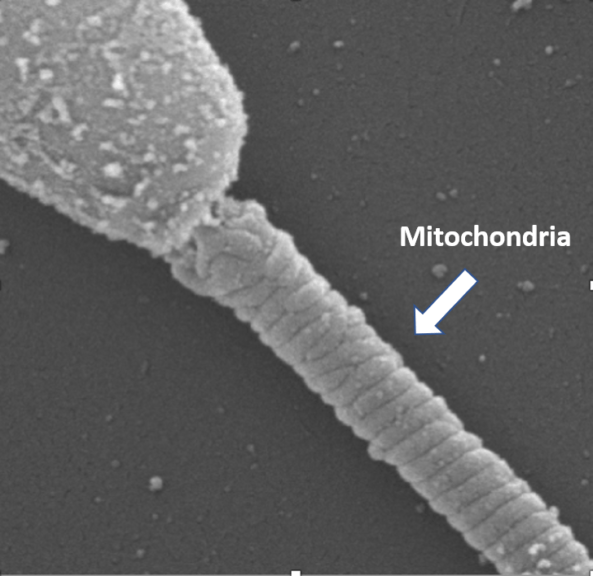
- Mammalian sperm cells contain 50-75 crescentic-shaped mitochondria with an end-to-end arrangement in its middle piece wrapping the outer dense fibers of the tail in a helical fashion. This arrangement of sperm mitochondria is known as a mitochondrial sheath.

Figure 9: Middle piece of the sperm showing helically arranged mitochondria. Image Credit: SBS - Myocytes contain a large number of mitochondria arranged between the myofibrils. A typical ventricular cardiac myocyte may contain up to 7,000 mitochondria occupying up to 35% of the cell volume.
- Mature erythrocytes are completely devoid of any mitochondria. Hence, they are dependent only on anaerobic respiration for their energy requirements.
- Fibroblasts contain several fused mitochondria often extending up to a length of 50 μm.
- Most eukaryotic organisms have mitochondria except for the Oxymonad monocercomonoides and Henneguya salminicola.
- Plant protoplasts usually contain several hundreds of mitochondria that are physically discrete and evenly distributed mitochondria due to cyclosis.
Which types of human cells are abundant in mitochondria? The mitochondria produce large amounts of energy through oxidative phosphorylation of organic molecules during cellular respiration. They are capable of using glucose and oxygen to produce energy (and releasing carbon dioxide and water in the process) for use in many metabolic processes. Thus, it is not surprising to find several mitochondria in high-energy-requiring cells, such as muscle cells.
Do all cells have mitochondria? There are cells that lack mitochondrion, such as mature red blood cells of mammals.
Dysfunction and Disease
So far, we have discussed enough to know why mitochondria are important for the survival and functioning of the cells. Therefore, any dysfunction in the mitochondria can also lead to disease. Mitochondrial dysfunction can arise from mutations in the mitochondrial genome, mutations in the nuclear genome that encodes essential mitochondrial components, or it may be due to the adverse effects of certain drugs.
Mitochondrial diseases
Below is a list of diseases that occur due to accumulated mutations in the mitochondrial genome.
- Leber’s hereditary optic neuropathy
- Leigh’s syndrome
- NARP syndrome
- Kearns Sayre syndrome
- CPEO: chronic progressive external ophthalmoplegia
- MELAS syndrome – it is the most common mitochondrial disorder
- Pearson’s syndrome
These diseases are inherited exclusively from the mother. As a rule, mitochondrial diseases tend to affect the brain, eyes, and skeletal muscles. This is so because of the high dependency of these tissues on mitochondria for their energy requirements.
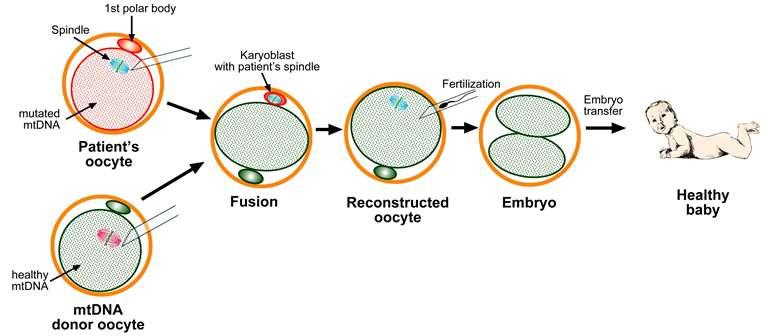
Mitochondrial replacement therapy
On 6th April 2016, the first male child was born using the Mitochondrial replacement therapy to a Jordanian couple in Mexico whose mother was suffering from Leigh’s syndrome. Mitochondrial replacement therapy is a novel IVF technique used when a woman with defects in her mitochondrial DNA intends to bear a genetic child. In this case, the cytoplasm with healthy mitochondrial DNA from a donor’s ovum is retained and the nuclear DNA from the intended mother is transferred leaving behind the defective mitochondrial genome. The hybrid ovum is then fertilized with the sperm of the father. The embryo created in this process contains healthy mitochondrial DNA from a donor mother and nuclear DNA from the intended mother & father.
Relationships to aging
The relationship of mitochondria with respect to aging is too complex and not fully understood. However, the generation of ROS, accumulations of mitochondrial mutations, and decreased functional capacity are mostly attributed to the aging process. Some possible mechanisms are discussed below.
- Generation of ROS – Mitochondria are the primary site of generation of reactive oxygen species (ROS). These ROS are capable of inducing damage to the macromolecules including DNA. The oxidative damage to the macromolecules has been associated with aging. The mutations induced in the DNA lead to defective functioning of the mitochondria further aggravating the release of ROS.
- Decreased respiratory capacity – With time decline in mitochondrial respiratory activity occurs. This alters cellular metabolism and contributes to aging. In addition to this, the decreased functional capacity is also responsible for the generation of more ROS.
- Mitophagy – Mitophagy is the piecemeal autophagy of the mitochondria. Mitophagy helps the cell to keep the number of dysfunctional mitochondria in check. With increasing age, mitophagy reduces, and the population of dysfunctional mitochondria increases.
- Mitochondrial mutations – Mitochondrial DNA is more prone to mutations than nuclear DNA as it does not have as efficient DNA repair mechanisms. With time these mutations can accumulate leading to the functional decline of mitochondria.
- Decline in Stem Cell Population – Stem cells are essential for maintaining the tissues in a healthy state. Mitochondria are known to regulate the function of stem cells. With increasing age decline in the stem cell population occurs.
- Cellular senescence – Mitochondria regulate cellular aging by regulating the metabolic profile of the cell. Profound changes in the cellular metabolome are causally associated with the senescent state of the cell.
- Mitochondrial unfolded protein response (UPRmt) – UPRmt is a cellular stress response triggered by mitochondrial stress that leads to misfolding of proteins in mitochondria. The nucleus senses this and upregulates the proteases & chaperones that get rid of the misfolded proteins. Decreased protease levels are associated with aging and age-related diseases such as cardiovascular disease and diabetes.
- DAMPs (damage-associated molecular patterns) – These are molecules released during stress. Mitochondrial DNA, one of the DAMPs is released in the blood during mitochondrial stress. The mitochondrial DNA levels in blood increase with age and it is associated with an age-related low-grade chronic inflammatory state.
- Mitochondrial derived peptides – Mitochondrial peptides such as humanin and MOTS-c can prevent Alzheimer’s disease and age-related insulin resistance respectively. They are also associated with an increase in longevity.
- Mitochondrial metabolism – Mitochondria produces NAD, which is a substrate for sirtuins. Sirtuins are proteins that play a role in longevity. With increasing age, the cellular levels of NAD keep on reducing.
Try to answer the quiz below to check what you have learned so far about mitochondria.

References
- Luo S. et al. (2018) Biparental inheritance of Mitochondrial DNA in Humans. PNAS, 115(51): 13039-13044 https://doi.org/10.1073/pnas.1810946115
- Romero-Garcia, S., & Romero-Garcia, S. (2019). Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). International Journal of Oncology, 54, 1155-1167. https://doi.org/10.3892/ijo.2019.4696
- Angajala, A., Lim, S., Phillips, J. B., Kim, J. H., Yates, C., You, Z., & Tan, M. (2018). Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Frontiers in immunology, 9, 1605. https://doi.org/10.3389/fimmu.2018.01605
- Wang, C., & Youle, R. J. (2009). The role of mitochondria in apoptosis*. Annual review of genetics, 43, 95–118. https://doi.org/10.1146/annurev-genet-102108-134850
- Ahlqvist K.J., Suomalainen, Hämäläinen R.H. (2015) Biochimica et Biophysica Acta (BBA) – Bioenergetics, 1847(11), 1380-1386. https://doi.org/10.1016/j.bbabio.2015.05.014
- Hirata, S., Hoshi, K., Shoda, T., & Mabuchi, T. (2002). Spermatozoon and mitochondrial DNA. Reproductive medicine and biology, 1(2), 41–47. https://doi.org/10.1046/j.1445-5781.2002.00007.x
- Dedkova, E. N., & Blatter, L. A. (2012). Measuring mitochondrial function in intact cardiac myocytes. Journal of molecular and cellular cardiology, 52(1), 48–61. https://doi.org/10.1016/j.yjmcc.2011.08.030
- Soltys B.J., Gupta R.S. (1992). Interrelationships of endoplasmic reticulum, mitochondria, intermediate filaments, and microtubules—a quadruple fluorescence labeling study. Biochemistry and Cell Biology, 70(10-11). https://doi.org/10.1139/o92-163
- Hamzelou J. (2016) Exclusive worlds first baby born with new 3 parent technique. https://www.newscientist.com/article/2107219-exclusive-worlds-first-baby-born-with-new-3-parent-technique/
- Bogenhagen D. F. (2012). Mitochondrial DNA nucleoid structure. Biochimica et biophysica acta, 1819(9-10), 914–920. https://doi.org/10.1016/j.bbagrm.2011.11.005
- Ho J. (2014). The regulation of apoptosis in kidney development: implications for nephron number and pattern?. Frontiers in pediatrics, 2, 128. https://doi.org/10.3389/fped.2014.00128
- Cooper G.M. The Cell A Molecular Approach. Eighth edition
©BiologyOnline.com. Content provided and moderated by Biology Online Editors.